Você sabe o que é a Síndrome de West?
- Benaia Silva

- 18 de abr. de 2021
- 7 min de leitura
Atualizado: 4 de jul. de 2021

Introdução
Epilepsia é um grupo de desordens neurológicas caracterizadas por descargas elétricas cerebrais anormais. Sua manifestação inclui perda de consciência, convulsões, espasmos, alterações sensoriais e distúrbios do sistema nervoso autônomo.
A síndrome de West é uma forma de epilepsia severa, que acomete crianças de até um ano de idade . Ela é caracterizada por uma tríade de sintomas que incluem: espasmos infantis, atraso de desenvolvimento neuropsicomotor e hipsarritmia (um padrão anormal de ondas cerebrais que aparecem no exame de eletroencefalograma).
Contexto histórico
Foi o médico William James West que descreveu essa síndrome pela primeira vez, no ano de 1841, quando decidiu escrever uma carta a respeito dos sintomas observados em seu filho para a revista científica The Lancet. Nessa carta, intitulada “On a peculiar form of infantile convulsions.” traduzido para o português ‘’Uma peculiar forma de convulsões infantis.’’, ele detalha a evolução dos espasmos observados na criança ao longo dos meses, que ora aconteciam em flexão, ora em extensão e que ocorriam repetidas vezes durante o dia, associado à um atraso no desenvolvimento tanto motor quanto intelectual.
Em 1951, Vasquez e Turner enquadraram essa síndrome na classificação das epilepsias e associaram os sintomas clínicos aos achados anormais no exame eletroencefalográfico. Mas foi só em 1960,na 9a edição do Colóquio de Marselha, que o neurologista francês Henri Jean Pascal Gastaut, R. G. Soulaysol e seus contribuintes deram a esse conjunto de manifestações o epônimo de síndrome de West.
Prevalência/ Incidência
A síndrome de West é um síndrome epiléptica rara, que acomete meninos com maior frequência do que meninas e tem uma incidência aproximada de 0,43 por 1000 nascidos vivos. Sua prevalência atinge valores de 0,15 a 0,20 casos por 1000 crianças abaixo de 11 anos de idade. Ela ocorre aproximadamente entre 3 a 12 meses de vida, tendo um pico de incidência aos 6 meses de vida.
Etiologia
A síndrome de West pode ser classificada quanto à etiologia (causa) em três categorias: sintomática, criptogênica e idiopática. Ela é categorizada em sintomática quando a causa da lesão cerebral é bem definida, podendo ser causada por malformações cerebrais, infecções, hemorragias, lesões hipóxico-isquêmicas, erros inatos do metabolismo e condições genéticas, como a esclerose tuberosa e a síndrome de Down. É classificada em criptogênica quando há alteração no exame neurológico, associado a uma suspeita de causa orgânica (causa física) e é idiopática quando não se pode delimitar uma causa, nesse caso o desenvolvimento motor pode estar normal. A descoberta da etiologia demanda uma anamnese detalhada, com uma análise da gravidez, do parto e da fase neonatal.
Manifestações Clínicas

A síndrome de West é um tipo raro de epilepsia caracterizada por crises epilépticas denominadas espasmos epilépticos, que podem ser de extensão ou flexão, sendo este último tipo o mais comum.
A apresentação clássica da síndrome de West consiste em episódios curtos de flexão abrupta do tronco e do pescoço e adução dos braços, esses espasmos tônicos são bilaterais, duram alguns segundos, são repetidos em salvas e ocorrem majoritariamente antes de dormir ou ao despertar. Durante as crises os olhos da criança podem permanecer fixos ou desviar (para o lado ou para cima), a criança pode fazer caretas ou piscar excessivamente, isso pode estar acompanhado de alterações cardíacas e respiratórias.
Após as crises é comum que as crianças apresentem irritabilidade ou sonolência. É importante ressaltar que a duração, a intensidade e o grupo muscular acometido durante as crises pode variar de criança para criança.
No eletroencefalograma observa-se hipsarritmia,um padrão caracterizado por ondas agudas multifocais de amplitude superior à 200 µV e por acentuada desorganização da atividade elétrica com descargas de espículas.
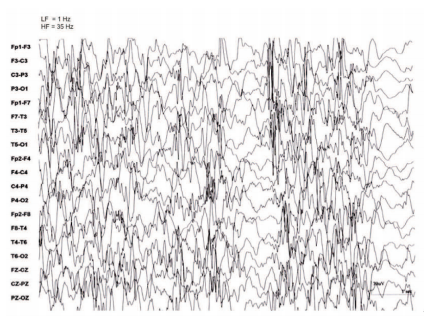
A maior parte das crianças portadoras desta síndrome podem ter desenvolvimento cognitivo deficitário e regressão ou atrasos na obtenção de habilidades que demandam coordenação de músculos e movimentos voluntários.
Diagnóstico
O diagnóstico da síndrome de West é feito a partir da obtenção de um relato detalhado do histórico de crises e da forma como elas se manifestaram. Esses detalhes são fornecidos pela pessoa que estava ao lado da criança quando as crises aconteceram, nesse sentido, vídeos do paciente no período dos espasmos são importantes, uma vez que existem outras condições não epilépticas (como cólicas, por exemplo), que acontecem na infância e podem ser confundidas com espasmos infantis.
A obtenção do histórico familiar e a realização de um exame físico geral detalhado, focado em obter uma avaliação neurológica completa (a fundoscopia deve estar incluída) também são parte da avaliação diagnóstica.
O exame eletroencefalograma é o principal a ser feito, pois através dele são verificados padrões anormais de atividade elétrica cerebral. Este é um exame indolor e não invasivo, onde são colocados eletrodos no couro cabeludo da criança, que captam as ondas cerebrais emitidas ora durante períodos de vigília (acordada), ora durante o sono. Crianças com esta síndrome podem necessitar de dois ou mais exames de eletroencefalograma, caso o primeiro exame não seja característico e o quadro clínico da criança for muito compatível com Síndrome de West. É essencial monitorar a atividade elétrica cerebral tanto em vigília quanto durante o sono. Se o padrão denominado Hipsarritmia for verificado, especialmente durante o sono, isso pode sugerir que a criança tem o diagnóstico de Síndrome de West.
A Tomografia Computadorizada de Crânio e a Ressonância Magnética são úteis para investigação da causa da Síndrome de West, ajudam na detecção de malformações cerebrais, calcificações e outros tipos de lesão causadoras dos espasmos epiléticos.
Se a causa da síndrome for devido a infecção, esta pode ser detectada por exames de sangue, urina e por punção lombar. Se a causa provável for de origem genética é importante realizar testagem genética, sendo o principal exame realizado o exoma.
Tratamento

O tratamento dessa síndrome visa melhorar a qualidade de vida, interrompendo o atraso de desenvolvimento que continua a ocorrer enquanto a síndrome não é tratada. As principais medicações utilizadas são hormônio adrenocorticotrófico (ACTH), Vigabatrina e Corticoide (principalmente prednisolona e metilprednisolona).
Atualmente a primeira linha de tratamento é uma terapia combinada feita com a vigabatrina associada ao ACTH intramuscular, que atuam reduzindo os espasmos e a promovendo melhora da hipsarritmia no exame eletroencefalográfico. Também como primeira linha de tratamento é possível fazer corticóide associado a vigabatrina, caso o ACTH esteja indisponível.
O ACTH promove redução ou interrupção total dos espasmos e melhora a Hipsarritmia em cerca de 50-75% dos pacientes em poucas semanas, tendo melhor resposta quando combinado a Vigabatrina.
Existe a alternativa de utilizar Vigabatrina isoladamente, porém este medicamento tem uma taxa de sucesso de cerca de 50% no tratamento da síndrome de West, e tem maior efetividade nos casos associados ao complexo de Esclerose Tuberosa. Inicialmente utilizam-se doses baixas e elas são aumentadas gradativamente, até chegar em altas doses. O principal efeito colateral do uso prolongado da Vigabatrina é o comprometimento visual causado por uma toxicidade na retina.
Pode-se também utilizar Prednisolona via oral dividida em quatro doses ao dia sob um regime de 6 em 6 horas, sendo recomendado associar a Vigabatrina para melhor resposta.
Além dos tratamentos farmacológicos é possível fazer dieta cetogênica, sendo esta rica em gorduras, com uma quantidade adequada de proteínas e pobre em carboidratos. Esta dieta deve ser feita sob supervisão de nutricionista e pediatra responsável pela criança. Contribui para melhora do quadro sindrômico, uma vez que reduz a fonte de energia necessária para que o cérebro gere a crise, no caso a glicose. A dieta baseada em gorduras gera como produto os corpos cetônicos, uma fonte alternativa de energia cerebral que não se relaciona aos fenômenos epilépticos.
A cirurgia é considerada o último recurso, ela consiste na remoção da área de anormalidade cortical em um procedimento denominado ressecção cortical. Ela é considerada a última alternativa para aqueles pacientes irresponsivos às terapias com ACTH e/ou Vigabatrina, Corticoide e dieta cetogênica. Pode ser realizada nos pacientes que apresentam anormalidades bem definidas na estrutura cerebral e este método de tratamento deve ser considerado após realização de um videoeletroencefalograma prolongado evidenciando que as crises são provenientes do local da lesão cerebral.
Prognóstico
O prognóstico depende muito da causa da síndrome, da idade de apresentação, da resposta ao tratamento e do tempo decorrido do início dos espasmos até o início da intervenção farmacológica.
Sabe-se que o prognóstico é mais favorável em casos que a etiologia é classificada em criptogênica ou idiopática, estes pacientes têm menor probabilidade de terem comprometimento cognitivo grave e de desenvolverem transtornos do espectro autista.
A idade de apresentação também influencia, uma vez que quando ela é superior a 4 meses, o prognóstico é melhor. Aproximadamente um terço das crianças com esta síndrome podem desenvolver crises epilépticas recorrentes e refratárias após o tratamento da Síndrome de West. Os pacientes podem evoluir para síndrome de Lennox-Gastaut, com variados tipos de crises epilépticas, que são de difícil controle, associadas à deficiência intelectual.
O outro terço dos pacientes continuará tendo espasmos epiléticos na idade adulta e o último terço pode ter espasmos que irão se resolver com o tempo, isso geralmente ocorre quando a etiologia não é clara. Foi verificado também que quanto menor o tempo até o início do tratamento e quanto mais sustentada a resposta à intervenção, melhor o prognóstico.
Orientação aos pais
É importante que os pais tentem filmar as crises para facilitar o diagnóstico pelo neuropediatra e afastar possíveis diagnósticos diferenciais. Existe uma ONG voltada para pessoas com deficiência, denominada ‘‘ Força do Bem’’, fundada em 2003 pela atriz Isabel Fillardis, cujo filho possui Síndrome de West. Essa ONG foi criada a fim de difundir conhecimento, auxiliar mães com poucas condições financeiras para cuidar das crianças portadoras de deficiência e servir como um espaço para que as famílias e pessoas detentoras dessa e de outras síndromes compartilhem suas experiências. A entidade desenvolveu o primeiro banco de dados brasileiro de pessoas com deficiência, que conta com mais de um milhão de cadastros, a fim de lutar por políticas públicas. Para mais informações acesse www.aforcadobem.org.br (link)
Para saber como ajudar a criança no momento das crises epilépticas clique na imagem abaixo.


Este texto foi produzido como atividade do Projeto Padrinho Med.
O projeto Padrinho Med foi idealizado pela médica Flávia Ju e tem objetivo de aproximar os médicos dos estudantes de medicina, promovendo troca de experiências e produção de conteúdo tanto científico quanto informativo à população.

Autora do texto: Laura Comeli Ordonho
Acadêmica do 7º período de medicina da PUC - Campinas

Coautora e revisora: Benaia Silva - médica neurologista pediátrica.
Para mais informações sobre a autora clique na imagem ao lado.
REFERÊNCIAS
RODRIGUES MM, Vilanova LCP. Tratado de Neurologia Infantil. 1a. Edição, Editora Atheneu, 2016.
West Syndrome. Nacional Organization for Rare Disorders, 2019. Disponível em: https://rarediseases.org/rare-diseases/west-syndrome/. Acesso em: 3 de Abril de 2021.
PAVONE, P. et al. West Syndrome: a comprehensive review. Neurological Sciences. 2020; 41(12): 3547–3562.
Infantile Spasm (West Syndrome). Medscape, 2019. Disponível em: https://emedicine.medscape.com/article/1176431-overview#a7. Acesso em: 3 de Abril de 2021.
TRENTO, S. S. M. Síndrome de West: um estudo bibliográfico. Intelletto, v.4, n. especial, 2019.
OSBORNE, J.P. et al. The Underlying etiology of infantile spasms (West Syndrome): Information from the International Collaborative Infantile Spasms Study (ICISS). Epilepsia, 2019;00: 1-9.
O’CALLAGHAN, F.J.K. et al. Safety and effectiveness of hormonal treatment versus hormonal treatm
ent with vigabatrin for infantile spasms (ICISS): a randomised, multicentre, open-label trial. The Lancet, v. 16, 2017.
BARBAROSSA E.P. et al. West Syndrome: Clinical Characteristics, Therapeutics, Outcomes and Prognosis. Electron J Gen Med. 2020;17(2):em 190.



Comentários